
Representing Molecules¶
What is a Molecule?¶
One of the greatest achievements in chemistry was the development of the valence model of chemistry, where a molecule is represented as atoms joined by semi-rigid bonds that can be single, double, or triple. This simple mental model has little resemblance to the underlying quantum-mechanical reality of electrons, protons and neutrons, yet it has proved to be a remarkably useful approximation of how atoms behave in close proximity to one another, and has been the foundation of chemical instruction for well over a century.
The valence model is also the foundation of modern chemical information systems. When a Computer Scientist approaches a problem, the first task is to figure out a datamodel that represents the problem to be solved as information. To the Computer Scientist, the valence model naturally transforms into a graph, where the nodes are atoms and the edges are bonds. Computer Scientists know how to manipulate graphs - mathematical graph theory and computer science have been closely allied since the invention of the digital computer.
There are atoms and space. Everything else is opinion.
—Democritus
However, the valence model of chemistry has many shortcomings. The most obvious is aromaticity, which quickly required adding the concept of a non-integral “aromatic” distributed bond, to the single/double/triple bonds of the simple valence model. And that was just the start - tautomers, ferrocenes, charged molecules and a host of other common molecules simply don’t fit the valence model well.
This complicates life for the computer scientist. As we shall see, they are the source of most of the complexity of modern cheminformatics systems.
Older systems: Connection Tables¶
Most of the early (and some modern) representations of molecules were in a connection table, literally, a table enumerating the atoms, and a table enumerating the bonds and which atoms each bond connected. Here is an example of connection-table (CTAB) portion of an MDL “SD” file (the data portion is not shown here):
MOLCONV
3 2 0 0 1 0 1 V2000
5.9800 -0.0000 -0.0000 Br 0 0 0 0 0 0
4.4000 -0.6600 0.8300 C 0 0 0 0 0 0
3.5400 -1.3500 -0.1900 C 0 0 0 0 0 0
1 2 1 0
2 3 1 0
This simple example illustrates most of the key features. The molecule has three atoms, two bonds, and is provided with three-dimensional (x,y,z) coordinates. MDL provides extensive documentation for their various CTFile formats if you are interested in the details.
Connection tables can capture the valence model of chemistry fairly well, but they suffer from three problems:
1. They are very inefficient, taking on the order of a dozen or two of bytes of data per atom and per bond. Newer line notations (discussed below) represent a molecules with an average of 1.2 to 1.5 bytes per atom, or 6-8 bytes per atom if coordinates are added.
2. Many suffered from lack of specificity. For example, since hydrogens are often not specified, there can be ambiguity as to the electronic state of some molecules, because the connection-table format does not explicitly state the valence assumptions.
3. Most mix the concept of connectivity (what the atoms are and how they are connected) with other data such as 2D and 3D coordinates. For example, if you had two different conformers of a molecule, most connection tables would require you to specify the entire molecule twice, even though the connection table is identical in both.
Line Notations: InChI, SMILES, WLN and others¶
A line notation represents a molecule as a single-line string of characters.
- WLN - Wisswesser Line Notation
WLN, invented by William J. Wisswesser in the early 1950’s, was the first comprehensive line notation, capable of representing arbitrarily complex molecules correctly and compactly.
1H = CH4 Methane 2H = CH3-CH3 Ethane 3H = CH3-CH2-CH3 Propane QVR BG CG DG EG FG = C7HCl5O2 PentachlorbenzoateWLN was the first line notation to feature a canonical form, that is, the rules for WLN meant there was only one “correct” WLN for any particular molecule. Those versed in WLN were able to write molecular structure in a line format, communicate molecular structure to one another and to computer programs. Unfortunately, WLN’s complexity prevented widespread adoption. The rules for correct specification of WLN filled a small book, encoding those rules into a computer proved difficult, and the rules for the canonicalization were computationally intractable.
- SMILES - Simplified Molecular Input Line Entry System
The best-known line notation today is SMILES. It was by Arthur and David Weininger in response to a need for a simpler, more “human accessible” notation than WLN. While SMILES is not trivial to learn and write, most chemists can create correct SMILES with just a few minutes training, and the entire SMILES language can be learned in an hour or two. You can read more details here. Here are some examples:
C methane CC ethane C=C ethene Oc1ccccc1 phenolSMILES, like WLN, has a canonical form, but unlike WLN, Weininger relied on the computer, rather than the chemist, to convert a non-canonical SMILES to a canonical SMILES. This important separation of duties was key to making SMILES easy to enter. (Read more about canonicalization below.)
- InChI
InChI is the latest and most modern of the line notations. It resolves many of the chemical ambiguities not addressed by SMILES, particularly with respect to stereo centers, tautomers and other of the “valence model problems” mentioned above.
You can read more about InChI at the Official Web Site, or on the Unofficial InChI FAQ page.
Canonicalization¶
A critical feature of line notations is canonicalization - the ability to choose one “blessed” representation from among the many. Consider:
OCC ethanol
CCO ethanol
Both of these SMILES represent the same molecule. If we could all agree that one of these was the “correct” or “canonical” SMILES for ethanol, then we would always store it the same way in our database. More importantly, if we want to ask, “Is ethanol in our database” we know that it will only be there once, and that we can generate the canonical SMILES for ethanol and look it up.
(Note that in theory one can create a canonical connection table, too, but it’s not as useful since informatics systems usually have trouble indexing BLOBs - large objects.)
Line Notation versus Connection Tables: A practical matter¶
Why are line notations preferred over connection-table formats? In theory, either could express the same information. But there are practical difference, mostly related to the complexity of “parsing” a connection table. If you know that the whole molecule is on one line of a file, it’s easy to parse.
Line notations are also very nice for database applications. Relational databases have datatypes that, roughly speaking, are divided into numbers, text, and “everything else”, also known as “BLOBs” (Binary Large OBjects). You can store line notations in the “text” fields much more easily than connection tables.
Line notations also have pragmatic advantages. Modern Unix-like systems (such as UNIX, Linux and Cygwin) have a number of very powerful “filter” text-processing programs that can be “piped” together (connected end-to-end) to perform important tasks. For example, to count the number of molecules containing aliphatic nitrogen in a SMILES file, I can simply:
grep N file.smi | wc
(grep looks for a particular expression, in this case N, and
prints any line that contains it, and wc (“word count”) counts the
number of words and lines.)
This is just a simple example of the power available via “script” programs using “filters” on Unix-like systems. Unix filters are much less useful for connection-table formats, because each molecule is spread over many lines.
Query Languages: SMARTS¶
In addition to a typographical way to represent molecules, we also need a way to enter queries about molecules, such as, “Find all molecules that contain a phenol.”
With text, we’re familiar with the concept of typing a partial word, such as “ford” to find “Henry Ford” as well as “John Hartford”. For chemistry, we can also specify partial structures, and find anything that contains them. For example:
| Query | Database | Matches? |
|---|---|---|
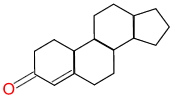 |
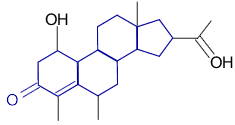 |
YES (matched portion highlighted in blue) |
|
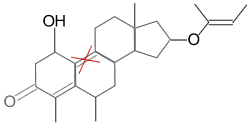 |
NO (double bond indicated doesn’t match) |

eMolecules is a one-stop shop for suppliers and information for over 8 million chemical compounds. Under the hood is a chemical registration technology based on Open Babel.
The simplest query language for chemistry is SMILES itself: Just
specify a structure, such as Oc1ccccc1, and search. This is how
eMolecules’ basic searching works (see Sidebar). It’s simple and, because of the
high-performance indexes in eMolecules, is also very fast.
However, for general-purpose cheminformatics, one needs more power.
What if the substructure you’re looking for isn’t a valid molecule?
For example ClccBr (1,2- substitution on an aromatic ring) isn’t a
whole molecule, since the concept of aromaticity is only sensible
in the context of a whole ring system.
Or what if the thing we’re looking for isn’t a simple atom such as Br, but rather a concept like “Halogen”? Or, “A terminal methyl”?
To address this, cheminformatics systems have special
query languages, such as SMARTS (SMiles ARbitrary Target
Specification). SMARTS is a close cousin to SMILES, but it has
expressions instead of simple atoms and bonds. For example, [C,N]
will find an atom that is either carbon or nitrogen.
IUPAC Names, Trade Names, Common Names¶
Chemistry also has three other important name systems:
- IUPAC Names
- IUPAC (the International Union of Pure and Applied Chemistry) established a naming convention that is widely used throughout chemistry. Any chemical can be named, and all IUPAC names are unambiguous. This textual representation is aimed at humans, not computers: Chemists versed in IUPAC nomenclature (which is widely taught) can read an IUPAC name and visualize or draw the molecule.
- Trade Names
- Names such as Tylenol™ and Valium™ are given to compounds and formulations by manufacturers for marketing and sales purposes, and for regulatory purposes.
- Common names
- Names such as “aspirin” or “alcohol” for substances that are in widespread use.